
Seeking principles
in the coordination among protein synthesis, metabolism, cell growth and differentiation
Research Projects
We aim to understand the rules governing emergent systems-level behavior and to use these rules to rationally engineer biological systems. We make quantitative measurements, often at the single-cell level, to test different conceptual frameworks and discriminate among different classes of models. These aims often motivate developing methods for sensitive quantification of protein levels, activities, modifications, synthsis and degradation rates. Areas of focus include:- Single-cell proteome biology
- Dynamics of protein synthsis and degradation
- RNA decoding and control of protein synthesis
- Resources for single-cell proteomics
Single-cell proteomics by mass spectrometry
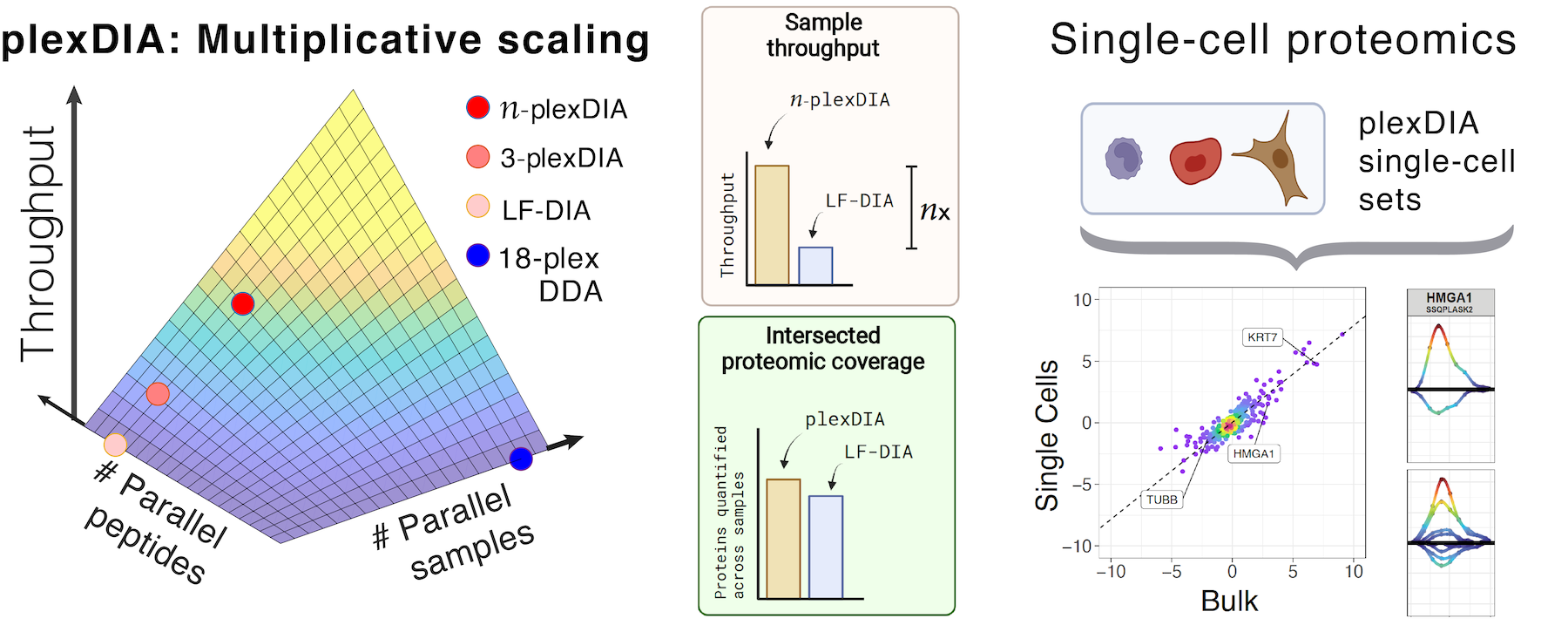
Many biological processes stem from the coordinated interactions of molecularly and functionaly diverse cells. However, this diversity is relatively unexplored at the proteome level because of the limitations of conventional affinity-based reagents for quantifying proteins in single cells. To alleviate these limitations, in 2017 our laboratory introduced Single Cell ProtEomics by Mass Spectrometry (SCoPE-MS). Since then, we developed more powerful and fully automated methods, including SCoPE2, pSCoPE, and plexDIA.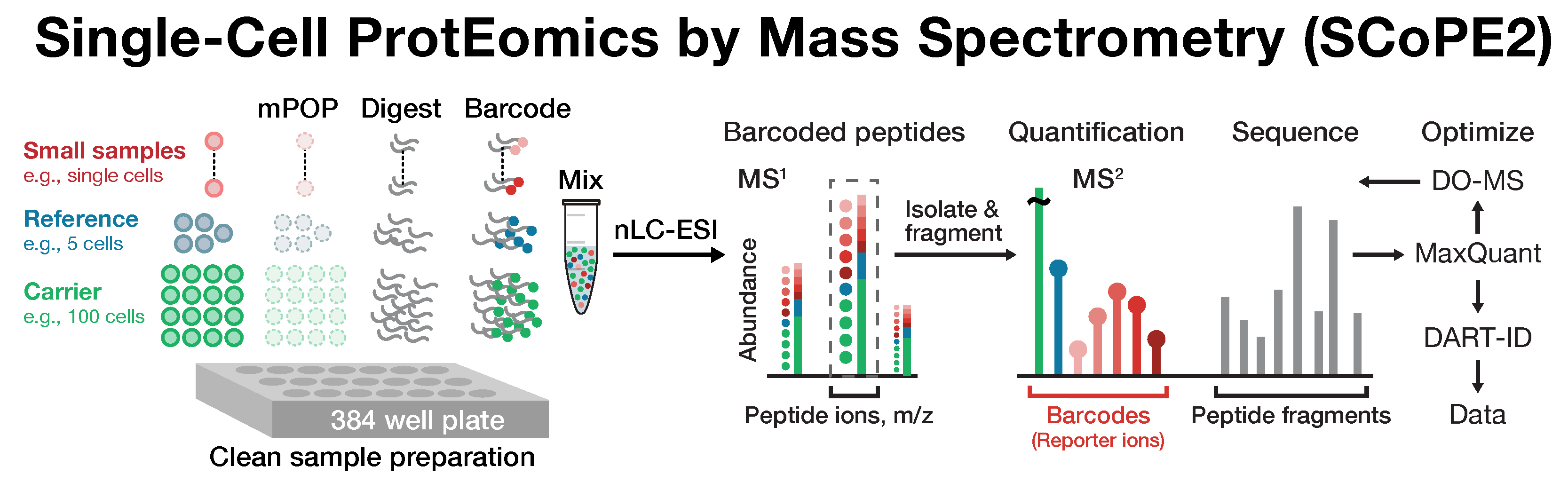
Taking advantage of ideas for advancing data acquisition and interpretation, we developed next generation methods that increase the sensitivity, data completeness and flexibility of single-cell protein analysis. These allow prioritization of thousands of proteins and highly parallel analysis of both single-cells and peptides. All of these methods can be implemented using accessible commercial equipment.
This research is funded by an Allen Distinguished Investigator Award, an iAward from Sanofi and an award from CZI seed networks. We collaborate with Parallel Squared Technology Institute (PTI), which was founded based on technologies developed by our team.

|

|
Dynamics of protein synthsis and degradation
Cellular protein concentrations are maintained through a balance of synthesis and clearance. Clearance occurs through both protein degradation and growth-dependent dilution. We have found that the impact of clearence in context-dependent and large, sometimes account for over 50 % of the measured variance in protein abundance. We study this contribution of protein degradation to shaping protein variation both across the proteome and across cell states.
Leduc A, Slavov N.✉ (2025)
Protein degradation and growth dependent dilution substantially shape mammalian proteomes
bioRxiv doi: 10.1101/2025.02.10.637566 | PDF | Github
Protein degradation and growth dependent dilution substantially shape mammalian proteomes
bioRxiv doi: 10.1101/2025.02.10.637566 | PDF | Github
Tracking proteome dynamics in single cells
One of the most significant recent developments in biomedical research and engineering is the explosion of technologies for quantifying thousands of genes in single cells. These technologies have transformed our ability to identify cell types and cell states in healthy and diseased human tissues. Our laboratory has contributed to these efforts by pioneering powerful methods for quantifying proteins in single cells at unprecedented scale. For the first time, we can examine single cells at systems level. Yet, these glimpses are static: we can only take one snapshot of a cell, showing us protein type and the amount of each protein, per cell. To overcome this limitation, we develop methods that will enable us to measure the abundance, the birth and the death rates of proteins across time. By introducing isotopically labeled amino acids at different time points over the life of a cell, we create a protein travelogue for each cell. Then, the travelogue of each cell is decoded by mass spectrometry, thus revealing the protein dynamics underpinning the life and functions of each cell. This technology can move biomedical research from the era of still pictures to the era of dynamic movies, the era when we can finally see and understand the dynamics of protein interactions that animate each cell.|
This research is funded by the Paul G. Allen Frontiers Group |

|
Alternate RNA decoding
Amino acid substitutions may substantially alter protein stability and function, but the con-tribution of substitutions arising from alternate translation (deviations from the genetic code) is unknown. We have identified 60,024 high confidence substitutions corresponding to 8,801 unique sites in proteins from 1,990 genes. Some substitutions are shared across samples, while others exhibit strong tissue-type and cancer specificity. Surprisingly, products of alternate translation are more abundant than their canonical counterparts for hundreds of proteins, suggesting sense codon recoding. Recoded proteins include transcription factors, proteases, signaling proteins, and proteins associated with neurodegenerative diseases. Mechanisms contributing to substitution abundance include protein stability, codon frequency, codon-anticodon mismatches, and RNA modifications. Some substitutions are shared across samples, while others exhibit strong tissue-type and cancer specificity. Surprisingly, products of alternate translation are more abundant than their canonical counterparts for hundreds of proteins, suggesting sense codon recoding. Recoded proteins include transcription factors, proteases, signaling proteins, and proteins associated with neurodegeneration. Mechanisms contributing to substitution abun-dance include protein stability, codon frequency, codon-anticodon mismatches, and RNA modifications. We characterize sequence motifs around alternatively translated amino acids and how substitution ratios vary across protein domains, tissue types and cancers. Both the sequence and the tissue-specificity of alternatively translated proteins are conserved between human and mouse. These results demonstrate the contribution of alternate translation to diversifying mammalian proteomes, and its association with protein stability, tissue-specific proteomes, and diseases.
Tsour S, Machne R, Leduc A, Widmer S, Slavov N.✉ (2024)
Alternate RNA decoding results in stable and abundant proteins in mammals
bioRxiv doi: 10.1101/2024.08.26.609665 | PDF | Web | Video
Alternate RNA decoding results in stable and abundant proteins in mammals
bioRxiv doi: 10.1101/2024.08.26.609665 | PDF | Web | Video
Ribosome-mediated translational regulation
All living cells must coordinate their metabolism, growth, division, and differentiation with their gene expression. Gene expression is regulated at multiple layers, from histone modifications (histone code) through RNA processing to protein degradation. While most layers are extensively studied, the regulatory role of specialized ribosomes (ribosome code) is largely unexplored. Such specialization has been suggested by the differential transcription of ribosomal proteins (RPs) and by the observation that mutations of RPs have highly specific phenotypes; particular RP mutations can cause diseases, known as ribosomopathies, and affect selectively the synthesis of some proteins but not of others. This selectivity and the differential RP transcription raise the hypothesis that cells may build specialized ribosomes with different stoichiometries among RPs as a means of regulating protein synthesis.While the existence of specialized ribosomes has been hypothesized for decades, experimental and analytical roadblocks (such as the need for accurate quantification of homologous proteins and their modifications) have limited the evidence to only a few examples, e.g., the phosphorylation of RP S6. We developed methods to clear these roadblocks and obtained direct evidence for differential stoichiometry among core RPs in unperturbed yeast and mammalian stem cells and its fitness phenotypes. We aim to characterize ribosome specialization and its coordination with gene regulation, metabolism, and cell growth and differentiation. We want to understand quantitatively, conceptually, and mechanistically this coordination with emphasis on direct precision measurements of metabolic fluxes, protein synthesis and degradation rates in absolute units, molecules per cell per hour.
|
This research is funded by the NIH Director's New Innovator Award |

|
Resources for Single-Cell Proteomics
- Single-cell proteomics networks
- Single-cell proteomics news
- Single-cell proteomics methods
- Single-cell proteomics data
Single-cell proteomics conference
1st Single-Cell Proteomics Conference, June 9 - 10, 2018
2nd Single-Cell Proteomics Conference, June 10 - 12, 2019
3rd Single-Cell Proteomics Conference, August 18 - 19, 2020
4th Single-Cell Proteomics Conference, August 16 - 18, 2021
5th Single-Cell Proteomics Conference, June 15 - 16, 2022
6th Single-Cell Proteomics Conference, June 1 - 2, 2023
7th Single-Cell Proteomics Conference, May 28 - 29, 2024
8th Single-Cell Proteomics Conference, May 27 - 29, 2025
|
Single-Cell Proteomics
Understand that cell
|