Research @ Slavov Lab
 |
We aim to understand the rules governing emergent systems-level physiological functions and to use these rules to rationally engineer biological systems.
We make direct quantitative measurements, often at the single-cell level, to test different conceptual frameworks and discriminate among different classes of models.
These aims often motivate developing methods for sensitive quantification of protein levels, activities, modifications, synthsis and degradation rates.
Areas of focus include:
Google Scholar |
PubMed |
ORCID |
CV |
bioRxiv |
arXiv |
Blog
While the existence of specialized ribosomes has been hypothesized for decades, experimental and analytical roadblocks (such as the need for accurate quantification of homologous proteins and their modifications) have limited the evidence to only a few examples, e.g., the phosphorylation of RP S6. We developed methods to clear these roadblocks and obtained direct evidence for differential stoichiometry among core RPs in unperturbed yeast and mammalian stem cells and its fitness phenotypes. We aim to characterize ribosome specialization and its coordination with gene regulation, metabolism, and cell growth and differentiation. We want to understand quantitatively, conceptually, and mechanistically this coordination with emphasis on direct precision measurements of metabolic fluxes, protein synthesis and degradation rates in absolute units, molecules per cell per hour.
This research is funded by a NIH Director's New Innovator Award.
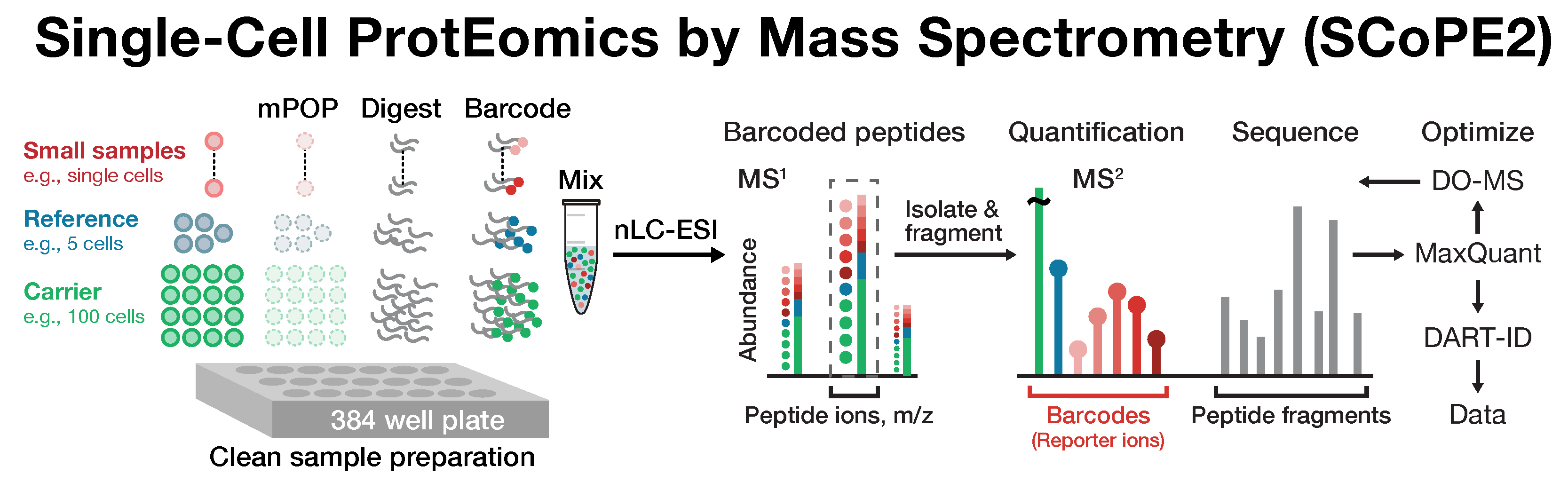
SCoPE2 introduced automated and miniaturized sample preparation that substantially lowers cost and hands-on time. It uses data-driven analytics to optimize instrument parameters for sampling more ion copies per protein, thus supporting quantification with improved count statistics. Furthermore, SCoPE2 uses peptide identification enhanced by incorporating retention time information within a principled framework, DART-ID.
SCoPE2 enables scalable, robust and affordable quantification of about 1,000 proteins per single cell, and over 3,000 proteins across many cells. This coverage is achieved with 1 hour of analysis time per SCoPE2 set (about 15 min / cell), which allowed us to analyze hundreds of cells on a single instrument in a couple of days. Importantly, SCoPE2 succeeded in delivering and quantifying hundreds of ion copies from most detected proteins. This observation strongly supports the feasibility of single-cell LC-MS/MS protein quantification without amplification.
Research Projects
Ribosome Specialization
All living cells must coordinate their metabolism, growth, division, and differentiation with their gene expression. Gene expression is regulated at multiple layers, from histone modifications (histone code) through RNA processing to protein degradation. While most layers are extensively studied, the regulatory role of specialized ribosomes (ribosome code) is largely unexplored. Such specialization has been suggested by the differential transcription of ribosomal proteins (RPs) and by the observation that mutations of RPs have highly specific phenotypes; particular RP mutations can cause diseases, such as cancer and Diamond Blackfan anemia, and affect selectively the synthesis of some proteins but not of others. This selectivity and the differential RP transcription raise the hypothesis that cells may build specialized ribosomes with different stoichiometries among RPs as a means of regulating protein synthesis.While the existence of specialized ribosomes has been hypothesized for decades, experimental and analytical roadblocks (such as the need for accurate quantification of homologous proteins and their modifications) have limited the evidence to only a few examples, e.g., the phosphorylation of RP S6. We developed methods to clear these roadblocks and obtained direct evidence for differential stoichiometry among core RPs in unperturbed yeast and mammalian stem cells and its fitness phenotypes. We aim to characterize ribosome specialization and its coordination with gene regulation, metabolism, and cell growth and differentiation. We want to understand quantitatively, conceptually, and mechanistically this coordination with emphasis on direct precision measurements of metabolic fluxes, protein synthesis and degradation rates in absolute units, molecules per cell per hour.
This research is funded by a NIH Director's New Innovator Award.
Single cell proteomics
Cellular heterogeneity is important to biological processes, including cancer and development. However, proteome heterogeneity is largely unexplored because of the limitations of existing methods for quantifying protein levels in single cells. To alleviate these limitations, our laboratory developed Single Cell ProtEomics by Mass Spectrometry (SCoPE-MS), and validated its ability to identify distinct human cancer cell types based on their proteomes. To further advance single-cell protein analysis, we developed SCoPE2.
SCoPE2 introduced automated and miniaturized sample preparation that substantially lowers cost and hands-on time. It uses data-driven analytics to optimize instrument parameters for sampling more ion copies per protein, thus supporting quantification with improved count statistics. Furthermore, SCoPE2 uses peptide identification enhanced by incorporating retention time information within a principled framework, DART-ID.
SCoPE2 enables scalable, robust and affordable quantification of about 1,000 proteins per single cell, and over 3,000 proteins across many cells. This coverage is achieved with 1 hour of analysis time per SCoPE2 set (about 15 min / cell), which allowed us to analyze hundreds of cells on a single instrument in a couple of days. Importantly, SCoPE2 succeeded in delivering and quantifying hundreds of ion copies from most detected proteins. This observation strongly supports the feasibility of single-cell LC-MS/MS protein quantification without amplification.